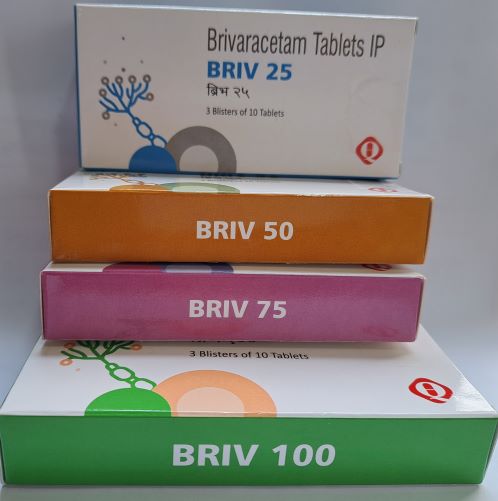
BRIV
Brivaracetam
Film-Coated Tablets
PRESENTATION:
Each Film Coated Tablet Contains Brivaracetam IP 25/ 50/75 & 100 mg
Packing: Blister
Pack Size: 3*10
CLINICAL PARTICULARS
Therapeutic Indications
Briv tablets is indicated as add-on therapy in the treatment of partial onset seizures with or without secondary generalization in patients from 4 years of age with epilepsy.
DOSE AND METHOD OF ADMINISTRATION
Adults
Briv at doses between 50 and 200 mg/day has been shown to be effective as adjunctive therapy in the treatment of partial onset seizures. Initial dose titration to an effective dose is not required for tolerability.
The daily dose is administered in two equally divided doses, once in the morning and once in the evening. The recommended starting dose as per clinical trials is 100 mg/day. In accordance with good prescribing practice, Briv may be initiated at a dose of 50 mg per day. Based on individual patient response, the dose may be adjusted between 50 mg/day and 200 mg/day in steps of 50 mg per day every 2 weeks. Briv may be taken with or without food.
Brivaracetam may be initiated with either intravenous or oral administration. When converting from oral to intravenous administration or vice versa, the total daily dose and frequency of administration should be maintained.
In accordance with current clinical practice, if Briv has to be discontinued, it is recommended to withdraw it gradually.
If patients missed one dose or more, it is recommended that they take a single dose as soon as they remember.
The film-coated tablets must be taken orally whole with liquid.
Use in Patients with Impaired Renal Function
The dose should be monitored in any form of renal impairment. Briv is not recommended in end stage renal disease patients undergoing dialysis due to lack of data.
Use in Patients with Impaired Hepatic Function
Exposure to brivaracetam was increased by 50%, 57% and 59% in adult patients with chronic liver disease belonging to Child-Pugh classes A, B and C, relatively to matched healthy controls. In adults, a 50 mg/day starting dose should be considered. In adolescents weighing 50kg or greater, a 50 mg/day starting dose is recommended. A maximum daily dose of 150 mg administered in 2 divided doses is recommended for all stages of hepatic impairment.
In pediatric patients weighing less than 50 kg, a 1 mg/kg/day starting dose is recommended. The maximum dose should not exceed 3 mg/kg/day.
Use in Elderly (65 Years and Older)
No dose reduction is necessary in elderly patients.
Use in Children
Adolescents weighing 50 kg or greater
The daily dose is administered in two equally divided doses, once in the morning and once in the evening. The recommended starting dose is 50 mg/day. Brivaracetam may also be initiated at 100 mg/day based on physician assessment of need for seizure control. The recommended maintenance dose is 100 mg/day. Based on individual patient response, the dose may be adjusted between the effective dose range of 50 mg/day and 200 mg/day.
Children from 4 years of age or adolescents weighing less than 50 kg
The daily dose is administered in two equally divided doses, once in the morning and once in the evening. The recommended starting dose is 1mg/kg/day. Brivaracetam may also be initiated at 2 mg/kg/day based on physician assessment of need for seizure control. The recommended maintenance dose is 2 mg/kg/day. Based on individual patient response, the dose may be adjusted between the effective dose range of 1 mg/kg/day and 4 mg/kg/day.
The physician should prescribe the most appropriate formulation and strength according to weight and dose.
There are insufficient data to recommend the use of Briv in children under 4 years of age (see Special Warnings and Precautions for Use).
CONTRAINDICATIONS
Hypersensitivity to the active substance or to any of the excipients.
SPECIAL WARNINGS AND PRECAUTIONS FOR USE
Suicidal Behavior and Ideation
Antiepileptic drugs, including Briv, increase the risk of suicidal thoughts or behavior in patients taking these drugs for any indication. Patients treated with any AED for any indication should be monitored for the emergence or worsening of depression, suicidal thoughts or behavior, and/or any unusual changes in mood or behavior.
Pooled analyses of 199 placebo controlled clinical trials (monotherapy and adjunctive therapy) of different AEDs showed that patients randomized to one of the AEDs had approximately twice the risk (adjusted relative risk 1.8, 95% CI: 1.2, 2.7) of suicidal thinking or behavior compared to patients randomized to placebo. In these trials, which had a median treatment duration of 12 weeks, the estimated incidence rate of suicidal behavior or ideation among 27,863 AED treated patients was 0.43%, compared to 0.24% among 16,029 placebo treated patients, representing an increase of approximately one case of suicidal thinking or behavior for every 530 patients treated. There were four suicides in drug treated patients in the trials and none in placebo treated patients, but the number is too small to allow any conclusion about drug effect on suicide.
The increased risk of suicidal thoughts or behavior with AEDs was observed as early as one week after starting drug treatment with AEDs and persisted for the duration of treatment assessed. Because most trials included in the analysis did not extend beyond 24 weeks, the risk of suicidal thoughts or behavior beyond 24 weeks could not be assessed.
Briv can cause hypersensitivity reactions. Bronchospasm and angioedema have been reported in patients taking Briv. If a patient develops hypersensitivity reactions after treatment with Briv, the drug should be discontinued. Briv is contraindicated in patients with a prior hypersensitivity reaction to brivaracetam or any of the inactive ingredients (see Contraindications).
Discontinuation
In accordance with current clinical practice, if Briv has to be discontinued it is recommended this be done gradually to minimize the potential for rebound seizures.
Use in Hepatic Impairment
There are limited clinical data on the use of brivaracetam in patients with pre-existing hepatic impairment. Dose adjustments are recommended for patients with hepatic impairment (see Dose and Method of Administration).
No adverse liver changes were seen in rats and monkeys following chronic administration of brivaracetam at exposure well above (up to 33-fold) the mean human exposure at the clinical dose of 200 mg/day. In dogs, brivaracetam administration resulted in adverse liver changes, mainly porphyria, at an exposure level close to mean human exposure at the clinical dose of 200 mg/day. However, toxicological data accumulated on brivaracetam and on a structurally related compound indicate that the dog liver changes have developed through mechanisms not relevant for humans.
Use in the Elderly
The three pivotal double blind placebo controlled studies included 38 elderly patients aged between 65 and 80 years. Although data are limited, the efficacy was comparable to younger subjects.
No dose adjustment is needed in elderly patients.
Pediatric Use
Briv is not recommended for use in children under 4 years of age as safety and efficacy has not yet been established in this population. Limited efficacy data is available from open label studies in children 1 month to < 16 years of age with partial onset seizures and other epilepsy syndromes.
The potential adverse effects of long-term oral administration of brivaracetam on neonatal growth and development were investigated in juvenile rats and dogs. In juvenile rats, the highest dose tested, 600 mg/kg/day, was associated with adverse developmental effects (i.e. mortality, clinical signs, decreased body weight, lower brain weight and non-reversible effects on auditory startle responses). There were no adverse neuropathological or brain histopathological findings. The NOAEL was considered to be 300 mg/kg/day. In juvenile dogs, the dose of 100 mg/kg/day induced adverse liver changes similar to those observed in adult animals. There were no adverse effects on growth, bone density or strength, brain and neurobehavioral assessments and neuropathology evaluation. Similar exposure to brivaracetam was achieved in adult vs. juvenile animals at the NOAEL, except at postnatal day 4 where higher exposure was achieved in juveniles compared to adults.
INTERACTIONS WITH OTHER MEDICINES AND OTHER FORMS OF
INTERACTIONS
Interaction studies have only been performed in adults.
Effects of other substances on brivaracetam
The hydrolysis of brivaracetam is mediated by non-CYP dependent amidase. The hydroxylation of brivaracetam appears to be a minor elimination pathway primarily mediated by CYP2C19 (see Pharmacological Properties). CYP-mediated oxidation is responsible for a limited portion of brivaracetam’s elimination.
Thus, co-administration with CYP inhibitors is unlikely to significantly affect brivaracetam exposure.
Co- administration with CYP450 strong inducer, rifampicin decreases brivaracetam plasma concentrations by 45%. Prescribers should consider increasing the brivaracetam dose in patients starting treatment with rifampicin and decreasing when stopping rifampicin therapy. Brivaracetam concentrations were not significantly modified by CYP3A inhibitors and CYP2C19 inhibitors. In vitro assays showed that brivaracetam disposition should not be significantly affected by any CYP (e.g. CYP1A, 2C8, 2C9, 2C19, 2D6 and 3A4) or transporter (e.g. P-glycoprotein, BCRP, MRPs) inhibitors.
Effects of Brivaracetam on Other Medicinal Products
Brivaracetam is not expected to cause clinically significant inhibition or induction of the clearance of other drugs metabolized by CYP450 isoforms. In vitro studies have shown that brivaracetam exhibits little or no inhibition of CYP450 isoforms at plasma concentrations achieved following a therapeutic dose. Brivaracetam did not induce CYP enzymes at therapeutic concentrations. Interaction studies to determine the potential inhibitory effects on transporters concluded that there were no clinically relevant effects.
Antiepileptic Drugs
Potential interactions between brivaracetam (50 mg/day to 200 mg/day) and other AEDs were investigated in a pooled analysis of plasma drug concentrations from all phase 2-3 studies and in a population exposure response analysis of placebo controlled phase 3 studies in adjunctive therapy in the treatment of partial onset seizures. The effect of the interactions on the plasma concentration is summarized as mentioned:
Carbamazepine 26% decrease
Phenobarbital 19% decrease
Phenytoin 21% decrease
Brivaracetam is a moderate reversible inhibitor of epoxide hydrolase resulting in an increased concentration of carbamazepine epoxide, an active metabolite of carbamazepine. In controlled studies, the carbamazepine epoxide plasma concentration increased by a mean of 37%, 62% and 98% with little variability at brivaracetam doses of 50 mg/day, 100 mg/day and 200 mg/day respectively. No toxicity was observed, however, if tolerability issues arise when co-administered, carbamazepine dose reduction should be considered.
Oral Contraceptives
Co- administration of brivaracetam (100 mg/day) with an oral contraceptive containing ethinylestradiol (0.03 mg) and levonorgestrel (0.15 mg) did not influence the pharmacokinetics of either substance.
When brivaracetam was co- administered at a dose of 400 mg/day (twice the recommended maximum daily dose) with an oral contraceptive containing ethinylestradiol (0.03 mg) and levonorgestrel (0.15 mg), a reduction in estrogen and progestin AUCs of 27% and 23%, respectively, was observed without impact on suppression of ovulation (no change was observed in the endogenous markers estradiol, progesterone, luteinizing hormone, follicle stimulating hormone, and sex hormone binding globulin). No study with lower doses of oral contraceptives has been performed.
Alcohol
Brivaracetam increased the effect of alcohol on psychomotor function, attention and memory in a pharmacokinetic and pharmacodynamics interaction study in healthy subjects. There was no pharmacokinetic interaction.
FERTILITY, PREGNANCY AND LACTATION
Effects on Fertility
No human data on the effect of brivaracetam on fertility are available. In rats, there was no adverse effect on male or female fertility following oral administration of brivaracetam at doses at least 15 times the maximal recommended human dose based on body surface area and plasma concentrations.
Use in Pregnancy
(Category B3)
There are no adequate data on the use of brivaracetam in pregnant women. Brivaracetam was used as adjunctive therapy in clinical studies and when used with carbamazepine, it induced a dose related increase in the concentration of an active metabolite, carbamazepine epoxide (see Interactions with Other Medicines and Other Forms of Interactions).
There is insufficient data to determine the clinical significance of this effect in pregnancy. There are no data on human placental transfer. In rats, brivaracetam was shown to readily cross the placenta. The potential risk for humans is unknown.
As a precautionary measure, brivaracetam should not be used during pregnancy unless clinically necessary (if the benefit to the mother clearly outweighs the potential risk to the fetus).
Discontinuation of antiepileptic treatments may result in exacerbation of the disease which could be harmful to the mother and the fetus. If a woman decides to become pregnant, the use of brivaracetam should be carefully re-evaluated.
Animal studies did not detect any teratogenic potential of brivaracetam in either the rat or the rabbit.
There were no adverse effects on embryo fetal development following oral administration of brivaracetam to rats during the period of organogenesis at doses up to 600 mg/kg/day (AUC exposure 25 times clinical exposure at the MRHD), or following intravenous administration of the brivaracetam metabolite ucb-107092-1 at doses up to 1000 mg/kg/day (plasma concentration at least 40 times the plasma Cmax in healthy or renally impaired subjects). In rabbits, adverse effects on embryo fetal development were not apparent at oral doses up to 120 mg/kg/day (AUC exposure 3 times clinical exposure at the MRHD) during organogenesis despite the presence of overt maternotoxicity.
Maternotoxic exposure at 6 times the clinical AUC at the MRHD resulted in increased post implantation loss, fewer live fetuses and reduced fetal bodyweight. The potential risk for humans is unknown.
Use in Lactation
Brivaracetam is excreted in human breast milk. A decision should be made whether to discontinue nursing or to discontinue brivaracetam, taking into account the benefit of the drug to the mother.
EFFECTS ON ABILITY TO DRIVE AND USE MACHINES
No studies on the effects on the ability to drive and use machines have been performed. Patients should be advised not to drive a car or to operate other potentially hazardous machines until they are familiar with the effects of brivaracetam on their ability to perform such activities, as brivaracetam treatment has been associated with somnolence and other CNS related symptoms.
ADVERSE EFFECTS (UNDESIRABLE EFFECTS)
Clinical Studies
In all controlled and uncontrolled trials in patients with epilepsy, 2388 subjects have received brivaracetam, of whom 1740 have been treated for ≥ 6 months, 1363 for ≥ 12 months, 923 for ≥ 24 months, 733 for ≥ 36 months and 569 for ≥ 60 months (5 years).
In pooled placebo controlled adjunctive therapy studies involving 1558 adult patients with partial onset seizures (1099 patients treated with brivaracetam and 459 treated with placebo), 68.3% of patients treated with brivaracetam and 62.1% of patients treated with placebo experienced adverse events.
The most frequently reported adverse reactions (> 10%) with brivaracetam treatment were: somnolence (14.3%) and dizziness (11.0%). They were usually mild to moderate in intensity.
Somnolence and fatigue were reported at a higher incidence with increasing dose. The types of adverse events reported during the first 7 days of treatment were similar to those reported for the overall treatment period.
The discontinuation rate due to adverse events was 6.0%, 7.4% and 6.8% for patients randomized to brivaracetam at respectively the dose of 50 mg/day, 100 mg/day and 200 mg/day and 3.5% for patients randomized to placebo. The adverse reaction most frequently resulting in discontinuation of brivaracetam therapy was dizziness.
Ear and Labyrinth Disorders
Vertigo
Eye Disorders
Vision blurred
Diplopia
Gastrointestinal Disorders
Nausea
Diarrhea
Vomiting
Constipation
Abdominal pain upper
Toothache
General Disorders and Administration Site Conditions
Fatigue
Irritability
Infections and infestations
Nasopharyngitis
Upper Respiratory Tract Infection
Influenza
Bacteriuria
Oral herpes
Injury, Poisoning and Procedural Complications
Fall
Excoriation
Head injury
Investigations
Weight decreased
Gammaglutamyltransferase increased
Weight increased
Metabolism and Nutrition Disorders
Decreased appetite
Hyponatraemia
Musculoskeletal Disorders
Myalgia
Back pain
Pain in extremity
Nervous System Disorders
Somnolence
Dizziness
Headache
Convulsion
Tremor
Balance Disorder
Memory impairment
Paraesthesia
Ataxia
Sedation
Psychiatric Disorders
Insomnia
Anxiety
Depression
Nervousness
Respiratory, Thoracic and Mediastinal Disorders
Cough
Dyspnea
Skin and Subcutaneous Tissue Disorders
Rash
Pruritus
Eczema
Pediatric Population
A long-term, uncontrolled, open-label safety study included children (from 4 years to less than 16 years) who continued treatment after completing the PK study (see Pharmacology) and children directly enrolled into the safety study. Children who directly enrolled received a brivaracetam starting dose of 1 mg/kg/day and depending on response and tolerability, the dose was increased up to 5 mg/kg/day by doubling the dose at weekly intervals. No child received a dose greater than 200 mg/day. For children weighing 50 kg or greater the brivaracetam starting dose was 50 mg/day and depending on response and tolerability, the dose was increased up to a maximum of 200mg/day by weekly increments of 50mg/day.
From the pooled open-label safety and PK studies in adjunctive therapy, 149 children with partial onset seizures in the age range of 4-<16 years of age have received brivaracetam, of whom 116 have been treated for ≥6 months, 104 for ≥12 months, 58 for ≥24 months, 20 for ≥36 months. The safety profile of brivaracetam observed in children was consistent with the safety profile observed in adults.
As has been seen in patients with epilepsy, behavioral adverse reactions were more common in children than in adults. The types of events were, in general, consistent with the established brivaracetam safety profile in adults. The majority of events were mild or moderate in intensity, were non-serious, and did not lead to dose reduction or discontinuation of study drug. An additional adverse reaction reported in children was psychomotor hyperactivity (4.7%). As these are uncontrolled data, definitive causality cannot be concluded.
Suicidal ideation was reported in 4.7 % of pediatric patients (more common in adolescents) compared with 2.4 % of adults and behavioral disorders were reported in 24.8 % of pediatric patients compared with 15.1 % of adults.
Other Adverse Reactions (< 1%)
Other adverse reactions with a lower incidence rate which are considered important are listed below:
Immune System Disorders: Hypersensitivity including bronchospasm and angioedema (Special Warnings and Precautions for Use).
Blood and Lymphatic System Disorders: Neutropenia.
Psychiatric Disorders: Aggression, agitation, psychotic disorder.
OVERDOSE
Symptoms
There is limited clinical experience with Briv overdose in humans. Somnolence and dizziness have been reported in a healthy subject taking a single dose of 1400 mg of Briv.
Management of Overdose
There is no specific antidote for overdose with Briv. Treatment of an overdose should include general supportive measures. Since less than 10% of brivaracetam is excreted in urine, hemodialysis is not expected to significantly enhance brivaracetam clearance.
PHARMACOLOGICAL PROPERTIES
Pharmacodynamics Properties
Mechanism of Action
Brivaracetam displays a high and selective affinity for synaptic vesicle protein 2A (SV2A) in the brain. Binding to SV2A is considered to be the primary mechanism for brivaracetam anticonvulsant activity, however, the precise mechanism by which brivaracetam exerts is anticonvulsant activity has not been fully elucidated.
Effects on QT interval
The effect of brivaracetam on QTc prolongation was evaluated in a randomized, double-blind, positive-controlled (moxifloxacin 400 mg) and placebo-controlled parallel group study of brivaracetam (150 mg/day and 800 mg/day in two daily intakes) in 184 healthy subjects. There was no evidence that brivaracetam prolongs the QT interval.
Seizure Frequency
A statistically significant correlation has been demonstrated between brivaracetam plasma concentration and seizure frequency reduction from baseline in confirmatory clinical studies in adjunctive treatment of partial onset seizures. The EC50 (brivaracetam plasma concentration corresponding to 50% of the maximum effect) was estimated to be 0.57 mg/L. This plasma concentration is slightly above the median exposure obtained after brivaracetam doses of 50 mg/day.
Further seizure frequency reduction is obtained by increasing the dose to 100 mg/day and reaches a plateau at 200 mg/day.
PHARMACOKINETIC PROPERTIES
Absorption
Brivaracetam is rapidly and completely absorbed after oral administration. Pharmacokinetics is dose proportional from 10 to 600 mg.
The median tmax for tablets taken without food is 1 hour (tmax range is 0.25 to 3 h). Co- administration with a high fat meal slowed down the absorption rate of brivaracetam while the extent of absorption remained unchanged. The extent of absorption of brivaracetam is unchanged by food.
Distribution
Brivaracetam is weakly bound (≤ 20%) to plasma proteins. The volume of distribution is 0.5 L/kg, a value close to that of the total body water.
Due to its favorable lipophilicity (log P) resulting in high cell membrane permeability, brivaracetam penetrates rapidly into the brain. Brivaracetam is rapidly and evenly distributed in most tissues. In rodents, the brain to plasma concentration ratio equilibrates rapidly, indicating fast brain penetration, and is close to 1, indicating absence of active transport.
Metabolism
Brivaracetam is primarily metabolized by hydrolysis of the amide moiety to form the corresponding carboxylic acid, and secondarily by hydroxylation on the propyl side chain. The hydrolysis of the amide moiety leading to the carboxylic acid metabolite (34% of the dose in urine) is supported by hepatic and extra hepatic amidase (E.C.3.5.1.4). In vitro, the hydroxylation of brivaracetam is mediated primarily by CYP2C19. In vivo, in human subjects possessing ineffective mutations of CYP2C19, production of the hydroxy metabolite is decreased 10-fold while brivaracetam itself is increased by 22% or 42% in individuals with one or both mutated alleles. Therefore, inhibitors of CYP2C19 are unlikely to have a significant effect on brivaracetam. The 3 metabolites are not pharmacologically active.
Excretion
Brivaracetam is eliminated primarily by metabolism and by excretion in the urine. More than 95% of the dose, including metabolites, is excreted in the urine within 72 hours after intake. Less than 1% of the dose is excreted in faces and less than 10% of brivaracetam is excreted unchanged in urine. The terminal plasma half-life (t1/2) is approximately 9 hours.
Pharmacokinetics in Special Patient Groups
Gender
There are no differences in the pharmacokinetics of brivaracetam by gender.
Renal Impairment
A study in subjects with severe renal impairment (creatinine clearance < 30 mL/min/1.73 m2 and not requiring dialysis) revealed that the plasma AUC of brivaracetam was moderately increased (+ 21%) relative to healthy controls, while the AUC of the acid, hydroxy and hydroxyacid metabolites were increased 3, 4, and 21-fold, respectively. The renal clearance of these nonactive metabolites was decreased 10-fold. Human exposure of the 3 metabolites, hydroxy, acid and hydroxyacid, at the maximum therapeutic dose of brivaracetam was sufficiently covered by levels achieved at the no observed adverse effect level (NOAEL) in repeated dose toxicity studies in animals, including for patients with severe renal impairment. The hydroxyacid metabolite did not reveal any safety concerns in nonclinical studies. Brivaracetam has not been studied in patients undergoing hemodialysis (see Dose and Method of Administration).
Hepatic Impairment
A pharmacokinetic study in subjects with hepatic cirrhosis (Child-Pugh grades A, B, and C) showed similar increases in exposure to brivaracetam irrespective of disease severity (50%, 57% and 59%), relative to matched healthy controls. Dose adjustments are recommended for patients with hepatic impairment (see Dose and Method of Administration). No clinical data are available in pediatric patients with hepatic impairment.
Elderly (Over 65 Years of Age)
In a study in elderly subjects (65 to 79 years old; with creatinine clearance 53 to 98 mL/min/1.73 m2) receiving Briv 400 mg/day in bid administration, the plasma half-life of brivaracetam was 7.9 hours and 9.3 hours in the 65 to 75 and > 75 years groups, respectively. The steady-state plasma clearance of brivaracetam was slightly lower (0.76 mL/min/kg) than in young healthy male subjects (0.83 mL/min/kg). No dose adjustment is required (see Dose and Method of Administration).
Pediatric Population (1 Month to 16 Years of Age)
In a pharmacokinetic study with 3-week evaluation period and weekly fixed 3-step up-titration using the brivaracetam oral solution, 99 subjects aged 1 month to <16 years were evaluated. Brivaracetam was administered at weekly increasing doses of approximately 1.0 mg/kg/day, 2.0 mg/kg/day, and 4.0 mg/kg/day. All doses were adjusted by body weight, and did not exceed a maximum of 50 mg/day, 100 mg/day, and 200 mg/day. At the end of the evaluation period, subjects may have been eligible for entry into a long-term follow-up study continuing on their last received dose (see Adverse Effects (Undesirable effects)). Plasma concentrations were shown to be dose proportional in all age groups.
Population pharmacokinetics modeling indicated that the dose of 2.0 mg/kg twice a day provides the same steady-state average plasma concentration as in adults receiving 100 mg twice daily. The estimated plasma clearance was 1.61 L/h, 2.18 L/h and 3.19 L/h for children weighing 20 kg, 30 kg and 50 kg, respectively. In comparison, plasma clearance was estimated at 3.58 L/h in adult patients (70 kg body weight). Currently, no clinical data are available in neonates.
Race
The pharmacokinetics of brivaracetam was not significantly affected by race (Caucasian, black/
African American, Asian, American Indian/ Alaska Native, Hispanic/ Latino) in a population pharmacokinetic modeling from epilepsy patients.
PRECLINICAL SAFETY DATA
Genotoxicity
Genotoxicity was evaluated in vitro in bacterial (Ames test) and mammalian cells (mouse lymphoma assay, chromosomal aberration test in CHO cells) and in vivo in rats (bone marrow micronucleus assay) and mice. Brivaracetam showed no evidence of mutagenicity or clastogenicity.
Carcinogenicity
In a carcinogenicity study in mice, oral administration of brivaracetam for 104 weeks increased the incidence of liver tumors (hepatocellular adenoma and carcinoma) in males at the two highest doses (550, 700 mg/kg/day). At the no effect dose (400 mg/kg/day), exposure (plasma AUC) was similar to clinical exposure at the MRHD. These findings are considered to result from a non -genotoxic mode of action linked to a phenobarbitone-like liver enzyme induction, a known rodent specific phenomenon.
In rats, oral administration of brivaracetam for 104 weeks (150, 230, 450, 700 mg/kg/day) resulted in an increased incidence of benign thymomas in females at the highest dose. At the no effect dose, exposure (plasma AUC) was about eight times the clinical exposure at the MRHD. The human relevance of the findings in rats is uncertain.
SPECIAL PRECAUTIONS FOR STORAGE
Store Briv film-coated tablets below 30°C.